「“毛”病专栏」Alstrom综合征
Alstrom综合征(Alström syndrome,ALMS)是一种罕见的常染色体单基因隐性遗传疾病,在人群中患病率小于1/1,000,000。该综合征的特征是从婴儿期开始的进行性多器官病变,表现为视网膜锥杆营养不良、感音神经性听力损失、肥胖、高胰岛素血症/2型糖尿病、扩张型心肌病、肺纤维化,以及肝功能和肾功能衰竭[1]。ALMS的致病基因主要是ALMS1。ALMS1位于染色体2p13,目前已鉴定出超过200种突变位点,主要发生在第8、10或16外显子,由插入/缺失引起的无义或错义突变,导致蛋白翻译提前终止[2]。近年来的一些研究表明,ALMS1蛋白位于中心体和基体的近端(图1),在维持初级纤毛功能中发挥作用,ALMS也越来越被认为是一类纤毛功能障碍相关疾病[3, 4]。
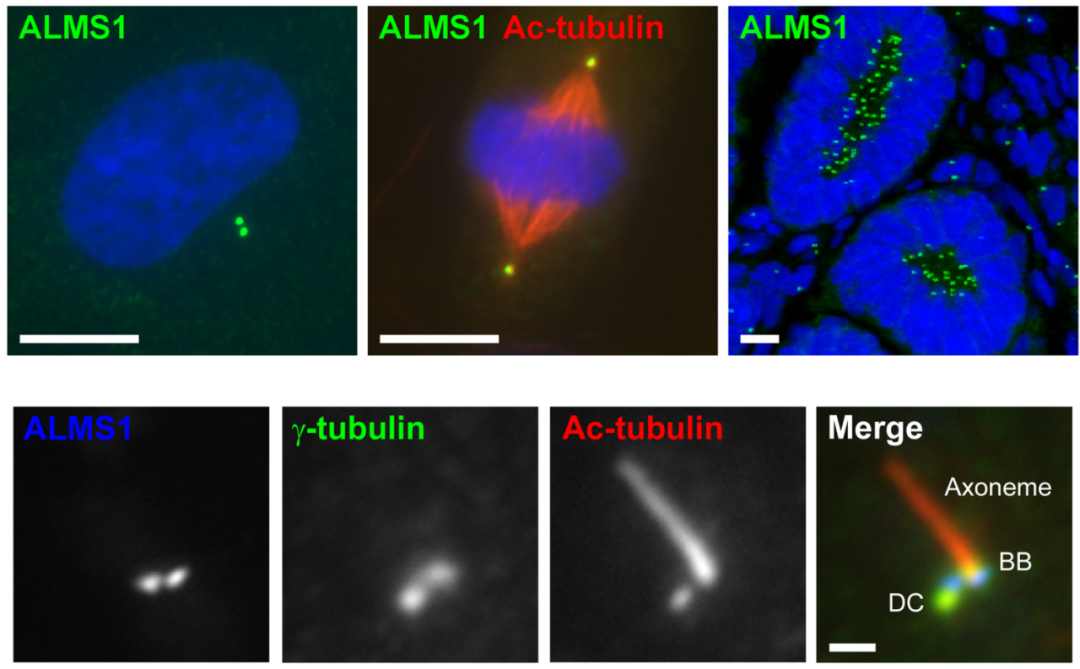
图1 ALMS1特异性定位于中心粒/基体的近端[2]。上图:通过免疫荧光实验,检测ALMS1在hTERT-RPE1细胞间期,有丝分裂中期以及人胎肾组织中的定位情况。下图:在hTERT-RPE1细胞中,检测ALMS1与γ-微管蛋白和中心粒/轴丝组分Ac-微管蛋白的定位。DC为子中心粒,BB为基体。
ALMS 临床诊断
ALMS临床表现复杂多变,即使在同一患病家族中临床表现也并不具有特异性。鉴于此,Marshall等建立了一套用于判断不同年龄阶段的临床诊断特征,有助于将ALMS与相关的疾病区分开来(表1)。
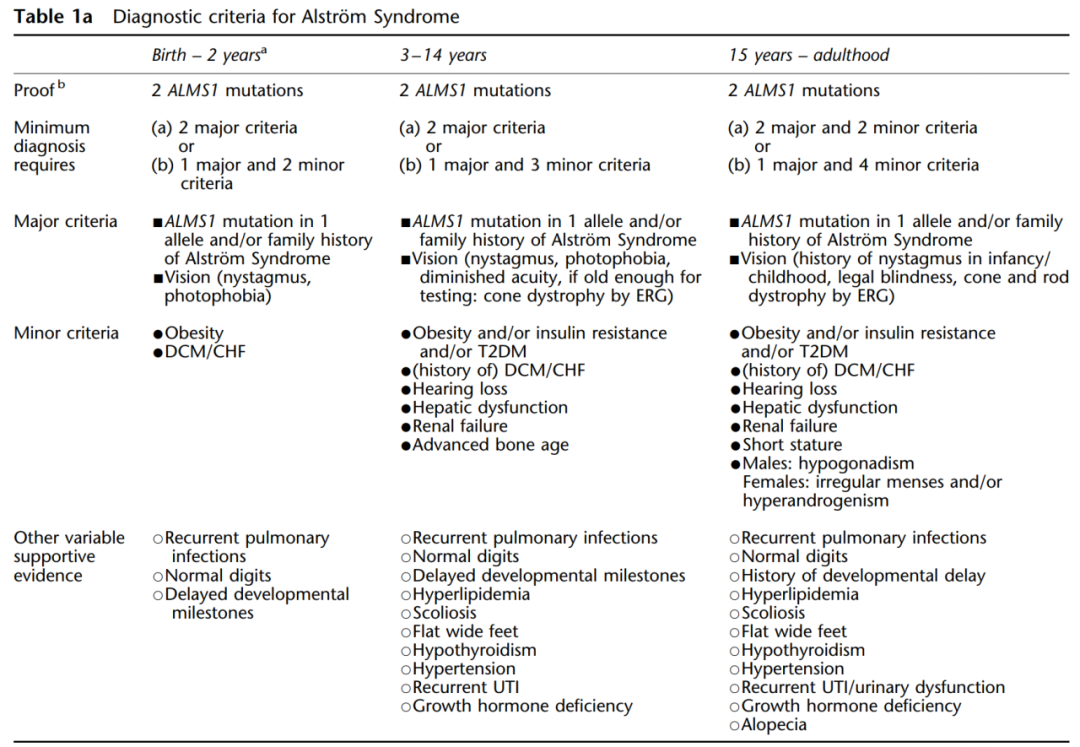
表1 ALMS在2岁以下婴儿、3~14岁儿童和15岁以上青少年/成人的临床和诊断特征[1]。
ALMS1基因的突变如何引起ALMS疾病?
对ALMS1的功能知之甚少,免疫荧光实验表明ALMS1主要定位于中心体和基体,提示其可能参与细胞内成熟的蛋白质向初级纤毛运输的过程(图2)。在ALMS1蛋白发生突变的细胞中,通常并不影响初级纤毛的发生,但可能通过影响纤毛内物质转运,从而影响在初级纤毛上的信号转导。
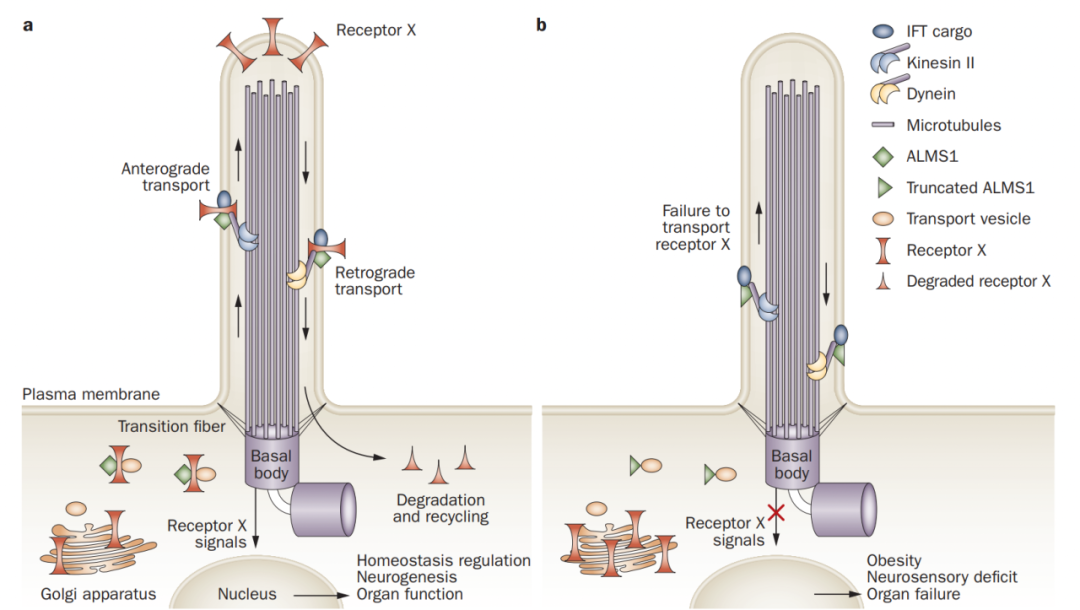
图2 ALMS1蛋白功能示意图[5]。a ALMS1蛋白可能参与细胞内从高尔基体运输一些受体到初级纤毛膜上。纤毛膜上的受体接收信号有助于调节细胞稳态、神经发生和器官的功能。b 截短突变的ALMS1未能将功能性蛋白运输到初级纤毛膜上,从而导致细胞信号传导缺陷。
ALMS 如何治疗?
目前还没有预防ALMS进行性器官受累的治疗方法,仅能给予支持性治疗。现有研究发现抗纤药物PBI-4050在Alstrom综合征中的具有抗炎、预防或逆转纤维化的作用[6]。希望随着研究的愈发深入,能够为我们提供更多针对性的治疗方法和有效的治疗药物。
内容:黄玉良
排版:罗玉婷
热门新闻
科室导诊| 看病不知道挂哪个科?对症找科室避免入坑!

涉及疫情传播风险!深圳培训机构被立案调查!南山1街道新增防范封控区

孩子,妈妈曾经也是个孩子

明码标价的卵子,毁掉多少女孩的一生

年纪轻轻腰老疼?原来是“它”在作祟……

