
概况
先天性角化不良(dyskeratosis congenita, DC),又称Zinsser-Cole-Engman综合征,是一种罕见的骨髓衰竭综合征,表现为显著的临床与遗传异质性。本病较为罕见,发病率约为1:1000000,无种族差异,可累及多个脏器系统,如皮肤、黏膜、眼、胃肠道、呼吸和血液系统等。目前,多认为本病是一种先天性遗传性疾病,以X-连锁隐性遗传方式为主,少数为常染色体显性遗传或常染色体隐性遗传,故男性患者明显多于女性[1]。
01
临床特点
DC的主要临床特点为皮肤网状色素沉着、甲营养不良和黏膜白斑的三联症症状。除此之外,患者亦可出现骨髓衰竭、智力发育迟缓、肺部疾患及泌尿生殖器、骨骼及胃肠道病变等。骨髓衰竭和肺部疾患为两大严重威胁生命的并发症。大约80%的患者并发骨髓衰竭,为本病的首要死因。三联症的主要表现形式为:
1.皮肤网状色素沉着:暴露部位出现网状皮肤异色病样改变,呈灰褐色斑疹,有时出现水疱或大疱,病情随着年龄增长而变化,皮损局限于面部、颈部和躯干上部,还会伴随皮肤起皱、萎缩、毛细血管扩张、多汗及掌跖角化过度等。
2.甲营养不良:指甲改变出现较早,通常发生在5~13岁之间,患者表现为甲营养不良或脱落,有时伴化脓性甲沟炎。
3.黏膜白斑:白斑可出现在任何黏膜部位,最常累及口腔黏膜、食管、尿道和泪管,导致口腔黏膜白斑、吞咽与排尿困难或泪溢。
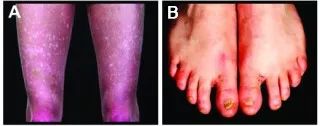
图1 先天性角化不良患者的临床表现
02
遗传学背景
截至目前,OMIM数据库和Orphanet数据库中已报道和DC相关的基因共12个,分别为TERC、TERT、NHP2、TINF2、NOP10、PARN、ACD、WRAP53、CTC1、USB1、RTEL1和DKC1。按照遗传方式可以分为:
1.X染色体隐性遗传的基因:DKC1
2.常染色体显性遗传的基因:TERC、TERT、TINF2、ACD、RTEL1
3.常染色体隐性遗传的基因:TERT、NOP10、NHP2、ACD、RTEL1、PARN、WRAP53、CTC1、USB1
其中TERT、ACD、RTEL1有常染色体显性遗传与常染色体隐性遗传两种遗传方式。
目前研究表明,端粒的缩短是引起先天性角化不良的主要原因。端粒酶对端粒长度的维持起着决定性的作用,端粒酶组分的多个基因突变均可引起端粒酶活性的改变。
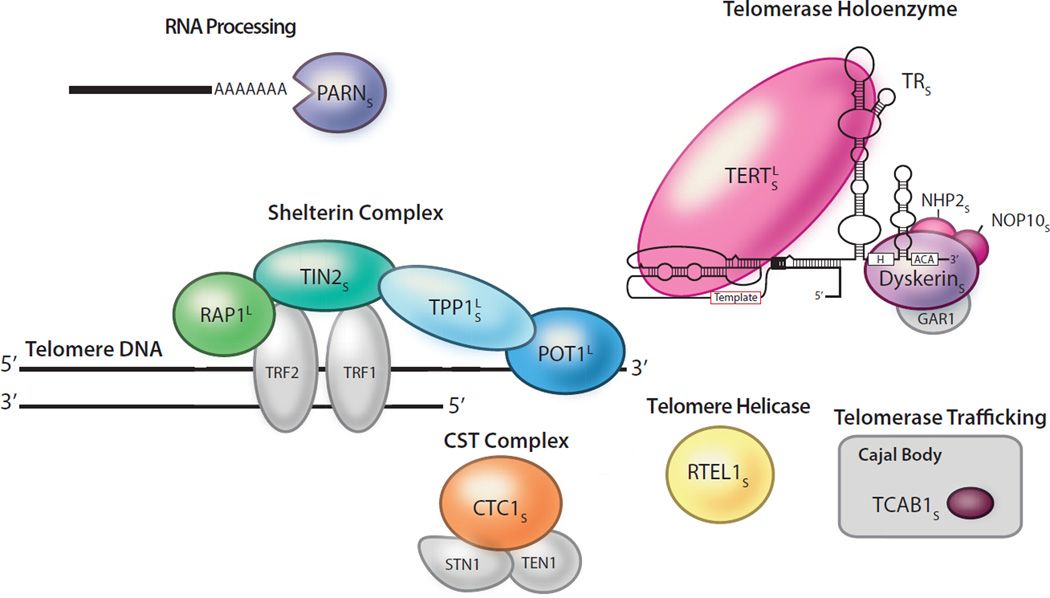
图2 不同基因与端粒酶的关系
突变体组分以颜色显示,灰色代表与疾病相关端粒组分的相关机制尚不明确。这些突变影响端粒酶催化活性或持续合成能力(TERT和TR),端粒酶生物合成(由DKC1编码的dyskerin),NOP10和NHP2或端粒酶运输(TCAB1也被称为WRAP53)。端粒综合征的突变也可能发生在shelterin组件中:TIN2(由TINF2编码),TPP1(由ACD编码),POT1,RAP1(由TERF2IP编码)。CTC1和RTEL1分别影响滞后链合成和端粒复制。PARN参与RNA加工和脱腺苷酸化[2]。
[参考文献]
[1] Wei L I, Xie X T, Paediatrics D O, et al. Diagnosis and treatment of the dyskeratosis congenita in children[J]. World Clinical Drugs, 2017.
[2] Stanley S E, Armanios M. The Short and Long Telomere Syndromes: Paired Paradigms for Molecular Medicine[J]. Current Opinion in Genetics & Development, 2015, 33:1.

